Alkohole beeinflussen das Schaltverhalten responsiver Polymere
Neuer Ansatz nutzt zeitaufgelöste Neutronenstreuung und theoretische Physik
2016-03-02 – Nachrichten aus dem Physik-Department
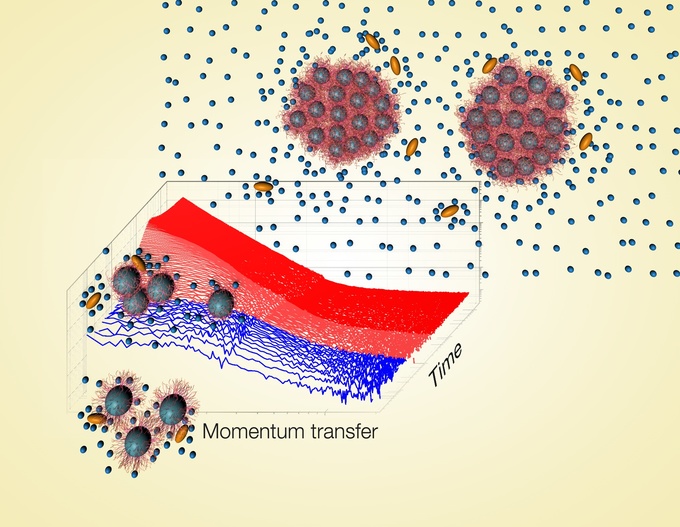
Die hintere Umschlagseite der aktuellen Ausgabe der Fachzeitschrift “Macromolecular Rapid Communications” zeigt Ergebnisse von Messungen an Polymer-Kolloiden aus einer Zusammenarbeit von Prof. Christine M. Papadakis vom Physik-Department der TUM mit Wissenschaftlerinnen und Wissenschaftlern der Universitäten Potsdam und Cambridge (Großbritannien) sowie vom Institut Laue-Langevin in Grenoble (Frankreich).
Kolloide aus fein verteilten, winzigen Teilchen in wässriger Lösung sind allgegenwärtig. So besteht zum Beispiel Kuhmilch aus einer Emulsion von kolloidalen Fetttröpfchen in Wasser. Als wasserbasierte Lacke oder Arzneimittel werden kolloidale Dispersionen technisch eingesetzt. Auch Makromoleküle wie Polymere, die in einer Flüssigkeit gelöst sind, können durch Assoziation Kolloide bilden. Um die Bildung der Kolloide für Anwendungen gezielt zu steuern, ist es wichtig, die zugrunde liegenden physikalischen Prozesse im Detail zu verstehen. Allerdings ist eine quantitative Beschreibung der Wechselwirkungspotentiale oft schwierig: Unter Umständen tragen nicht nur die komplexen Strukturen der die Kolloide bildenden Moleküle bei, sondern es müssen auch die komplexen Wechselwirkungen des Lösungsmittels berücksichtigt werden, was vor allem in wässriger Lösung eine wichtige Rolle spielt.
Faszinierendes Verhalten der Polymerteilchen bei einem Temperatursprung
In einer internationalen Zusammenarbeit haben Physikerinnen und Physiker des Physik-Departments die Aggregation von Polymeren an Poly(N-isopropylacrylamide) (PNIPAM), untersucht. Unterhalb der Trübungstemperatur von ~32 °C ist PNIPAM in Wasser löslich, also hydriert. Oberhalb dieser Temperatur ändert das Polymer seine Konfiguration von einem hydrierten Zustand in einen dehydrierten, es kollabiert und bildet große Aggregate. (Daher die Bezeichnung “thermoresponsiv”: Das Polymer reagiert deutlich auf geringe Temperaturänderungen.) Da diese Eigenschaft für mögliche Anwendungen in der Biomedizin und Mikrofluidik von großem Interesse ist, ist PNIPAM gegenwärtig eines der am intensivsten untersuchten thermoresponsiven Polymere. Hier interessiert insbesondere die Charakterisierung und Quantifizierung der Aggregation, die wesentlich von der Wechselwirkungsenergie zwischen den Makromolekülen abhängt.
Die vorliegende Studie untersucht selbstassemblierte Mizellen aus Blockcopolymeren, die aus PNIPAM und einem kurzen hydrophoben (wasserabstoßenden) Block eines anderen Polymers (Polystyrol, PS) bestehen. Da PNIPAM sich bei Zimmertemperatur im Gegensatz zu PS gut in Wasser löst, bilden sich kugelförmige Mizellen, in denen die Hülle aus hydriertem PNIPAM die PS-Kerne der Mizellen vom Lösungsmittel abschirmt. Die mizellaren Lösungen wurden in Wasser oder in Mischungen aus Wasser und Methanol bzw. Ethanol (zwei unterschiedlich große Alkohole) präpariert. Eine besondere Probenumgebung erlaubt schnelle Temperatursprünge über die Trübungstemperatur. Der damit induzierte Aggregationsprozess wurde mittels zeitaufgelöster Neutronen-Kleinwinkelstreuung untersucht, und zwar am Instrument D22 des Instituts Laue-Langevin (ILL) in Grenoble (Frankreich).
“Nur die hervorragende Probenumgebung, die das ILL bietet, erlaubt es, derart schnelle Temperaturänderungen durchzuführen und damit den Aggregationsvorgang mit hoher Zeitauflösung zu beobachten”, erläutert Christine M. Papadakis. Die Messungen zeigen, dass sich das Wachstum der Aggregate beschleunigt, je größer die zugefügten Alkoholmoleküle sind.
Maßgeschneiderte Eigenschaften von Kolloiden
Die Datenauswertung fand im Team mit Prof. Alessio Zaccone statt, der jüngst von der TUM an die Universität Cambridge wechselte. Er hat kürzlich ein theoretisches Modell entwickelt, das die Wachstumsrate von Aggregaten mit deren Wechselwirkungsenergie verknüpft. Es stellt sich heraus, dass die Anwesenheit der Alkoholmoleküle die Wechselwirkungenergie signifikant ändert. Dies ermöglicht es, durch den Zusatz von Alkohol die Selbstaggregation gezielt zu steuern und damit thermoresponsive Systeme an die jeweilige Anwendung anzupassen.
“Die Alkoholmoleküle stören die gut strukturierte Hydrathülle der hydrophilen Teile der Aggregatoberfläche. Damit reduziert sich die Abstoßung, was zur Beschleunigung des Wachstums der Aggregate führt”, erklärt Alessio Zaccone.
Das Zusammenwirken von modernen experimentellen und theoretischen Methoden führt hier zu einem besseren Verständnis der molekularen Wechselwirkungen in responsiven wässrigen Lösungen und erlaubt die Kontrolle ihres Schaltverhaltens. Aus grundsätzlichen Erwägungen kann der beschriebene Ansatz auch für andere Systeme verwendet werden, beispielsweise bei der Aggregation von Biomolekülen.
- Redaktion
- Dr. Johannes Wiedersich
Veröffentlichung
Links
Kontakt
- Prof. Christine Papadakis,
- Physik-Department, Technische Universität München,James-Franck-Str. 1, 85747 Garching, GermanyTel.: (+49) 89 289 12 447, Fax: (+49) 89 289 12 473E-Mail: papadakis@tum.de